IRB Types of Review
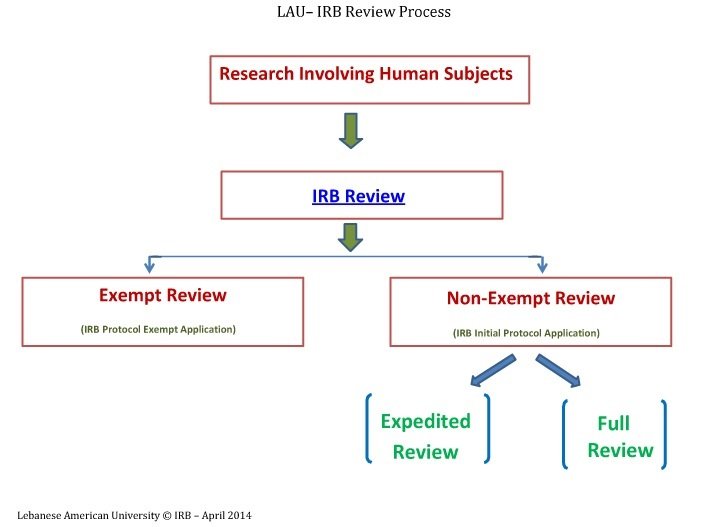
Exempt, Expedited, Full IRB Review
The IRB follows three distinct review types while reviewing research involving human subjects. These types are based on the regulations governing research and relate to the degree of risk to research subjects. For Full and Expedited review, please use the Initial Protocol Application and for Exempt review, please use the Protocol Exempt Application. You can refer to the explanation below and also you can also check the Investigator’s Manual - Policies and Procedures.
Exempt Review
Some human subject research might fall under an Exempt review process. In order to assess if your research project fits one of the exemption criteria, please see list below or 45CFR 46 101 (b) at http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html. The IRB office will make the final determination if your research project falls under an Exempt Review.
Research may be exempt from review when the only involvement of human subjects in the research falls into one of the following categories:
- Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as (i) research on regular and special education instructional strategies, or (ii) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods.
- Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior unless: (i) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects’ responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, or reputation. (However, when a study involves children being interviewed, questioned or surveyed, that study must be reviewed by the IRB and may not be exempt. Similarly, studies involving children and observation of public behavior in which the Principal Investigator (or other investigator) participates in the activities being observed must be reviewed by the IRB)
- Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures (e.g. anonymous questionnaire), interview procedures, or observation of public behavior that is not otherwise exempt if: (i) the human subjects are elected or appointed public officials or candidates for public office; or (ii) federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter.
- Research, involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects.
- Research and demonstration projects which are conducted by or subject to the approval of department or agency heads, and which are designed to study, evaluate, or otherwise examine: (i) public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives to those programs or procedures; or (iv) possible changes in methods or levels of payment for benefits or services under those programs.
- Taste and food quality evaluation and consumer acceptance studies, (i) if wholesome foods without additives are consumed or (ii) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the federal health authority or food inspection agency
Expedited Review
Some human subject research might fall under an Expedited Review process. The categories for Expedited review process are noted below and can be found at http://www.hhs.gov/ohrp/policy/expedited98.html
Applicability
- Research activities that (1) present no more than minimal risk to human subjects, and (2) involve only procedures listed in one or more of the following categories, may be reviewed by the IRB through the expedited review procedure authorized by 45 CFR 46.110 and 21 CFR 56.110. The activities listed should not be deemed to be of minimal risk simply because they are included on this list. Inclusion on this list merely means that the activity is eligible for review through the expedited review procedure when the specific circumstances of the proposed research involve no more than minimal risk to human subjects.
- The categories in this list apply regardless of the age of subjects, except as noted.
- The expedited review procedure may not be used where identification of the subjects and/or their responses would reasonably place them at risk of criminal or civil liability or be damaging to the subjects= financial standing, employability, insurability, reputation, or be stigmatizing, unless reasonable and appropriate protections will be implemented so that risks related to invasion of privacy and breach of confidentiality are no greater than minimal.
- The expedited review procedure may not be used for classified research involving human subjects.
- Investigators are reminded that the standard requirements for informed consent (or its waiver, alteration, or exception) apply regardless of the type of review—expedited or convened—utilized by the IRB.
Research Categories
- Clinical studies of drugs and medical devices only when condition (a) or (b) is met.
- Research on drugs for which an investigational new drug application (21 CFR Part 312) is not required. (Note: Research on marketed drugs that significantly increases the risks or decreases the acceptability of the risks associated with the use of the product is not eligible for expedited review.)
- Research on medical devices for which (i) an investigational device exemption application (21 CFR Part 812) is not required; or (ii) the medical device is cleared/approved for marketing and the medical device is being used in accordance with its cleared/approved labeling.
- Collection of blood samples by finger stick, heel stick, ear stick, or venipuncture as follows:
- from healthy, nonpregnant adults who weigh at least 110 pounds. For these subjects, the amounts drawn may not exceed 550 ml in an 8 week period and collection may not occur more frequently than 2 times per week; or
- from other adults and children2, considering the age, weight, and health of the subjects, the collection procedure, the amount of blood to be collected, and the frequency with which it will be collected. For these subjects, the amount drawn may not exceed the lesser of 50 ml or 3 ml per kg in an 8 week period and collection may not occur more frequently than 2 times per week.
- Prospective collection of biological specimens for research purposes by noninvasive means.
Examples: (a) hair and nail clippings in a nondisfiguring manner; (b) deciduous teeth at time of exfoliation or if routine patient care indicates a need for extraction; (c) permanent teeth if routine patient care indicates a need for extraction; (d) excreta and external secretions (including sweat); (e) uncannulated saliva collected either in an unstimulated fashion or stimulated by chewing gumbase or wax or by applying a dilute citric solution to the tongue; (f) placenta removed at delivery; (g) amniotic fluid obtained at the time of rupture of the membrane prior to or during labor; (h) supra- and subgingival dental plaque and calculus, provided the collection procedure is not more invasive than routine prophylactic scaling of the teeth and the process is accomplished in accordance with accepted prophylactic techniques; (i) mucosal and skin cells collected by buccal scraping or swab, skin swab, or mouth washings; (j) sputum collected after saline mist nebulization. - Collection of data through noninvasive procedures (not involving general anesthesia or sedation) routinely employed in clinical practice, excluding procedures involving x-rays or microwaves. Where medical devices are employed, they must be cleared/approved for marketing. (Studies intended to evaluate the safety and effectiveness of the medical device are not generally eligible for expedited review, including studies of cleared medical devices for new indications.)
Examples: (a) physical sensors that are applied either to the surface of the body or at a distance and do not involve input of significant amounts of energy into the subject or an invasion of the subject=s privacy; (b) weighing or testing sensory acuity; (c) magnetic resonance imaging; (d) electrocardiography, electroencephalography, thermography, detection of naturally occurring radioactivity, electroretinography, ultrasound, diagnostic infrared imaging, doppler blood flow, and echocardiography; (e) moderate exercise, muscular strength testing, body composition assessment, and flexibility testing where appropriate given the age, weight, and health of the individual. - Research involving materials (data, documents, records, or specimens) that have been collected, or will be collected solely for nonresearch purposes (such as medical treatment or diagnosis).
(NOTE: Some research in this category may be exempt from the HHS regulations for the protection of human subjects. 45 CFR 46.101 (b)-(4). This listing refers only to research that is not exempt.) - Collection of data from voice, video, digital, or image recordings made for research purposes.
- Research on individual or group characteristics or behavior (including, but not limited to, research on perception, cognition, motivation, identity, language, communication, cultural beliefs or practices, and social behavior) or research employing survey, interview, oral history, focus group, program evaluation, human factors evaluation, or quality assurance methodologies. (NOTE: Some research in this category may be exempt from the HHS regulations for the protection of human subjects. 45 CFR 46.101(b)-(2) and (b)-(3) This listing refers only to research that is not exempt.)
- Continuing review of research previously approved by the convened IRB as follows:
- where (i) the research is permanently closed to the enrollment of new subjects; (ii) all subjects have completed all research-related interventions; and (iii) the research remains active only for long-term follow-up of subjects; or
- where no subjects have been enrolled and no additional risks have been identified; or
- where the remaining research activities are limited to data analysis.
- Continuing review of research, not conducted under an investigational new drug application or investigational device exemption where categories two (2) through eight (8) do not apply but the IRB has determined and documented at a convened meeting that the research involves no greater than minimal risk and no additional risks have been identified.
In addition, expedited review is appropriate for minor changes in protocols and consent forms proposed for previously approved research during the period (one year or less) for which approval is authorized. Changes affecting risk, benefit, discomfort, or subject protections are not “minor” changes. Minor modifications include, for example, administrative changes to the protocol, changes to add follow-up calls when gathering initial data by telephone, or certain changes in the scheduling of medications.
Full IRB Review
Human subject research that does not fit any of the Expedited or Exempt review categories will require a Full IRB review at a convened meeting. The IRB meets once a month or as requested by the IRB chairman. Research applications are placed on the agenda and will be discussed at the next scheduled meeting. The IRB chairman might cancel a full IRB meeting if
- There are insufficient number of applications to be discussed,
- Inability to secure a quorum,
- University holiday; or
- Other reasons that may arise that makes a meeting unnecessary or inappropriate
The IRB uses a primary reviewer system for full IRB review. Application materials are sent to the IRB members scheduled to attend a meeting at least one week in advance of the meeting. Two members are selected by the chair, one as the primary reviewer and the other as a secondary reviewer for a research project.
The primary reviewer leads the discussion of each project at the full IRB convened meeting. The secondary reviewer adds any other relevant comments or clarifications. The members determine whether the project meets the criteria for approval or whether revisions to the study design are required. The Informed Consent Document is reviewed for accuracy, clarity, and inclusion of required and optional elements of consent. During the meeting, voting is by show of hands. By a majority of those present at the meeting, each project is either: (1) approved as submitted; (2) approved pending receipt of required minor revisions to study procedures, Informed Consent Document(s), or other written materials; (3) tabled pending review at a subsequent full board meeting after receipt of significant additional information or revisions, or (4) disapproved.
Written minutes of each full IRB meeting include: (1) attendance, (2) the number of votes to approve, table, disapprove, or abstain (without individual identification), (3) the basis for requiring changes in or disapproving the research, (4) the length of time until the next review, and (5) a summary of the discussion of controverted issues and their resolution.