
The Institutional Review Board (IRB) at the Lebanese American University (LAU) is responsible for reviewing and approving all research projects involving human subjects. This includes studies conducted at LAU, the LAU Medical Center–Rizk Hospital (LAUMC–RH), and the LAU Medical Center–Saint John’s Hospital (LAUMC–SJH), as well as any external research conducted by LAU faculty, students, or staff.
The primary mission of the IRB is to ensure that all proposed research complies with established ethical standards, including the core principles outlined in the Belmont Report, and aligns with national and international regulations and guidelines for the protection of human research participants.
IRB Meeting Dates & Deadlines for Submissions
Below is a list of meetings dates and deadlines for submission of research project that require Full Board Review at a specified and scheduled IRB meeting date.
Projects that fall under Exempt or Expedited review category are reviewed under expedited review procedure and not during the Full Board Review. Please check the review category for your study.
| IRB Meeting Dates Scheduled 2026 | Deadlines for Research Project Submission |
|---|---|
| January 9, 2026 Cancelled | December 9, 2025 |
| February 27, 2026 | January 27, 2026 |
| March 27, 2026 | February 27, 2026 |
| April 24, 2026 | March 24, 2026 |
| May 22, 2026 | April 22, 2026 |
| June 19, 2026 | May 19, 2026 |
| July 17, 2026 | June 17, 2026 |
IRB Membership Roster
As of October 2024
| Member Name | Title and Affiliation | Term of Appointment | Member Designation |
|---|---|---|---|
| Joseph Stephan, PhD |
Associate Professor, Gilbert & Rose-Marie Chagoury School of Medicine Director, IRB office, LAU |
September 2016 -present
February 2019 - present |
Scientific member
Chairperson, IRB
|
| Vanda Abi Raad, MD, MHPE, EMBA |
Associate Dean for Faculty Affairs and Development Clinical Professor and Chair for the Department of Anesthesiology Gilbert & Rose-Marie Chagoury School of Medicine, LAU |
June 2017 - present | Vice Chair IRB, Scientific member |
| Aniella Abi Gerges, PhD | Associate Professor, School of Medicine | December 2019 - present | Scientific member |
| Grace Dagher, PhD | Associate Professor, School of Business - LAU | December 2019 - present | Scientific member |
| Maha Habre, DNP, RN, CEN |
Clinical Assistant Professor, School of Nursing, LAU |
October 2020 - present | Scientific member |
| Abeer Hani, MD |
Assistant Professor and Pediatric Neurologist, Gilbert & Rose-Marie Chagoury School of Medicine, LAU |
January 2016 - Present | Scientific member |
| Fr. Ramy Wannous | Saint George Church, Broummana | October 2023 – present | Non-Scientific and Not affiliated member |
| Maya Zeineddine, PhD | Clinical Associate Professor, School of Pharmacy, LAU | October 2024 - present | Scientific member |
| Rami Smayra, LLM | Smayra Law Office | May 2019 - present | Non-scientific and not affiliated member |
| Pia Tohme, PhD | Assistant Professor, Psychology Department, School of Arts & Sciences, LAU | February 2022 - Present | Scientific member |
| Rana Rizk, PhD | Assistant Professor, Natural Sciences Department, School of Arts & Sciences, LAU | October 2022 - Present | Scientific member |
| Leen Baddour | Student ad hoc non voting member. School of Arts & Sciences – Psychology Student | October 2024 - Present | Non-scientific |
| Sally Sarkissian | Student ad hoc non voting member. School of Pharmacy – MS in Pharmaceutical Development and Management Student | October 2024 - Present | Non-scientific |
| Karmen Baroudy, M.Sc., CIM | IRB Lead Program Manager, LAU | February 2012 - present |
Scientific, Non-voting member |
Submission Requirements
1. Overall Requirements
All research involving human subjects under the jurisdiction of the Lebanese American University and its affiliates (LAU Medical Center - Rizk Hospital & LAU Medical Center - Saint John’s Hospital) must be submitted to the LAU Institutional Review Board (IRB) for review and approval prior to commencing the research. This page provides you with easy checklists and processes for submission. Please refer also to the Investigator’s Manual - Policies and Procedures.
LAU IRB does not review research conducted by non LAU/LAUMCRH/LAUMCSJH personnel.
If a study involves extramural funding such as external funding agencies or sponsors, clearance must be secured from the relevant department at LAUMC–RH / LAUMC-SJH / LAU before submission to the IRB. Please contact the IRB office / or the GSR office for further details. For funding opportunities, please visit the Sponsored Program website
Please note the following critical points to be included in any research protocol for the IRB to assess
- Purpose and Rational of the study
- Targeted participant population and justification
- Sample size justification
- Manner of recruitment and informed consent
- Study procedures
- Anticipated risks and potential benefits to participants
- Steps taken to protect participants including management of adverse events.
2. LAU IRB Submission Checklist and Summary Presentation
The following information provides you with relevant information on what you need to submit as part of your LAU IRB Submission. For the Applications, Forms and Supporting Documents click here.
- IRB Submission Checklist for Faculty and Staff (click here)
- IRB Submission Checklist for Students (click here)
- IRB Submission Presentation (click here)
3. LAU IRB Submission process
For any funding queries, please contact the GSR office at gsrinfo@lau.edu.lb for more information.
4. LAU IRB Submission Fee Schedule
The IRB charges a fee for review of research studies involving human subjects for sponsored or funded research projects, as detailed below. Payment is expected independent of the IRB decision and is due within 30 days of receipt of invoice. For any questions, please contact the IRB office by email at irb@lau.edu.lb.
Effective February 2011, the fee schedule for LAU IRB review is as follows:
| Initial Submission - Pharmaceutical Sponsored Studies | $1500 plus VAT |
| Annual Continuing Review Submission - Pharmaceutical Sponsored Studies | $500 plus VAT |
| Initial Submission - Extramural Funding Agencies | $200 plus VAT |
| Annual Continuing Review Submission - Extramural Funding Agencies | $50 plus VAT |
5. IRB Review Timelines - Summary Table
| Type of Review* | Timelines** | Required Application*** |
|---|---|---|
| Full Review |
Review will take at least 30-45 business days from date of submission (complete submission must be at least 2 weeks prior to the next scheduled meeting) |
|
| Expedited Review | Review will take 20 business days from date of complete submission |
|
| Exempt Review | Review will take up to 10 business days from date of complete submission |
|
* Detailed explanation for the different Types of Review can be found under Guidance Documents.
** Timelines noted are for complete submissions. If a submission is missing documents, the submission will be returned to the PI with clarifications. Timelines also vary if there are any major concerns to the project submitted.
*** All submissions must include a completed and signed application along with all required documents as noted at the beginning of each application.
*** Whenever required, all investigators on the application must complete and sign the Investigator Financial Disclosure Form.
Applications, Forms, and Supporting Documents
Signature pages and IRB Applications
1. New Mandatory Automated Cover Form (RPSF) and Signature Pages, as applicable:
a. Research Proposal Submission Form (RPSF)
This form is only required for submissions by a Faculty: Not applicable for student
projects.
This form includes an option if the study is to be conducted at LAUMCRH & LAUMCSJH
b. Other required Signature Pages, as applicable
- LAU Simulation Center Signature Page (If the research is to take place at the LAU Simulation Center)
c. Adding LAUMCRH & LAUMCSJH as a research site after you have secured LAU IRB approval, please use the following form
2. IRB Applications
| ONLY One application should be submitted depending on the type of submission for review: | |
|
Initial Protocol Application - Biomedical Research (For all research projects that require Full or Expedited review) |
|
|
Initial Protocol Application – Social and Behavioral Research (For all research projects that require Full or Expedited review) |
|
|
Protocol Exempt Application (For all research projects that fall under an Exempt review) |
|
| Supplemental Sheet - Study Personnel | |
|
Classroom Project Application (For student projects in Research Methodology Courses) |
|
| Other required applications, as applicable and required, during the course of the Research Project and after Initial review and approval: | |
| Addition of Research Personnel to an already IRB Approved research study | |
|
Continuing Review Application (For all research projects that have been approved under Full or Expedited review and requires renewal to continue past expiry date noted in the Approval letter) |
|
|
Protocol Amendment Application (For all research projects where the PI intends to amend the protocol, informed consent, any change to the research project procedures) |
|
|
Request for Protocol Closure Form (For all research projects that have completed, closed, suspended or terminated) |
Reporting Forms
| Investigator Financial Disclosure Form | |
|
SAE/ Non-SAE Unexpected and Related Reporting Form (For Adverse Events occurring at the site) |
Supporting Guidance Documents and Templates
IRB Review Process
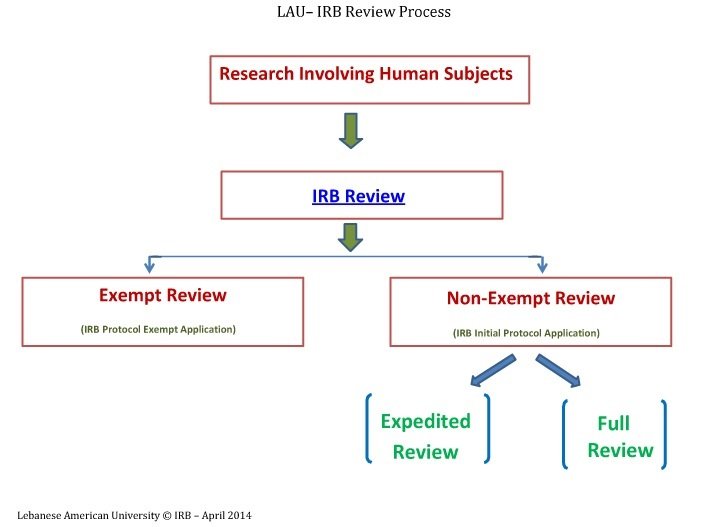
Exempt, Expedited, Full IRB Review
The IRB follows three distinct review types while reviewing research involving human subjects. These types are based on the regulations governing research and relate to the degree of risk to research subjects. For Full and Expedited review, please use the Initial Protocol Application and for Exempt review, please use the Protocol Exempt Application. You can refer to the explanation below and also you can also check the Investigator’s Manual - Policies and Procedures.
Exempt Review
Some human subject research might fall under an Exempt review process. In order to assess if your research project fits one of the exemption criteria, please see list below or 45CFR 46 101 (b) at http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html. The IRB office will make the final determination if your research project falls under an Exempt Review.
Research may be exempt from review when the only involvement of human subjects in the research falls into one of the following categories:
- Research conducted in established or commonly accepted educational settings, involving normal educational practices, such as (i) research on regular and special education instructional strategies, or (ii) research on the effectiveness of or the comparison among instructional techniques, curricula, or classroom management methods.
- Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior unless: (i) information obtained is recorded in such a manner that human subjects can be identified, directly or through identifiers linked to the subjects; and (ii) any disclosure of the human subjects’ responses outside the research could reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, or reputation. (However, when a study involves children being interviewed, questioned or surveyed, that study must be reviewed by the IRB and may not be exempt. Similarly, studies involving children and observation of public behavior in which the Principal Investigator (or other investigator) participates in the activities being observed must be reviewed by the IRB)
- Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures (e.g. anonymous questionnaire), interview procedures, or observation of public behavior that is not otherwise exempt if: (i) the human subjects are elected or appointed public officials or candidates for public office; or (ii) federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter.
- Research, involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects.
- Research and demonstration projects which are conducted by or subject to the approval of department or agency heads, and which are designed to study, evaluate, or otherwise examine: (i) public benefit or service programs; (ii) procedures for obtaining benefits or services under those programs; (iii) possible changes in or alternatives to those programs or procedures; or (iv) possible changes in methods or levels of payment for benefits or services under those programs.
- Taste and food quality evaluation and consumer acceptance studies, (i) if wholesome foods without additives are consumed or (ii) if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the federal health authority or food inspection agency
Expedited Review
Some human subject research might fall under an Expedited Review process. The categories for Expedited review process are noted below and can be found at http://www.hhs.gov/ohrp/policy/expedited98.html
Applicability
- Research activities that (1) present no more than minimal risk to human subjects, and (2) involve only procedures listed in one or more of the following categories, may be reviewed by the IRB through the expedited review procedure authorized by 45 CFR 46.110 and 21 CFR 56.110. The activities listed should not be deemed to be of minimal risk simply because they are included on this list. Inclusion on this list merely means that the activity is eligible for review through the expedited review procedure when the specific circumstances of the proposed research involve no more than minimal risk to human subjects.
- The categories in this list apply regardless of the age of subjects, except as noted.
- The expedited review procedure may not be used where identification of the subjects and/or their responses would reasonably place them at risk of criminal or civil liability or be damaging to the subjects= financial standing, employability, insurability, reputation, or be stigmatizing, unless reasonable and appropriate protections will be implemented so that risks related to invasion of privacy and breach of confidentiality are no greater than minimal.
- The expedited review procedure may not be used for classified research involving human subjects.
- Investigators are reminded that the standard requirements for informed consent (or its waiver, alteration, or exception) apply regardless of the type of review—expedited or convened—utilized by the IRB.
Research Categories
- Clinical studies of drugs and medical devices only when condition (a) or (b) is met.
- Research on drugs for which an investigational new drug application (21 CFR Part 312) is not required. (Note: Research on marketed drugs that significantly increases the risks or decreases the acceptability of the risks associated with the use of the product is not eligible for expedited review.)
- Research on medical devices for which (i) an investigational device exemption application (21 CFR Part 812) is not required; or (ii) the medical device is cleared/approved for marketing and the medical device is being used in accordance with its cleared/approved labeling.
- Collection of blood samples by finger stick, heel stick, ear stick, or venipuncture as follows:
- from healthy, nonpregnant adults who weigh at least 110 pounds. For these subjects, the amounts drawn may not exceed 550 ml in an 8 week period and collection may not occur more frequently than 2 times per week; or
- from other adults and children2, considering the age, weight, and health of the subjects, the collection procedure, the amount of blood to be collected, and the frequency with which it will be collected. For these subjects, the amount drawn may not exceed the lesser of 50 ml or 3 ml per kg in an 8 week period and collection may not occur more frequently than 2 times per week.
- Prospective collection of biological specimens for research purposes by noninvasive means.
Examples: (a) hair and nail clippings in a non-disfiguring manner; (b) deciduous teeth at time of exfoliation or if routine patient care indicates a need for extraction; (c) permanent teeth if routine patient care indicates a need for extraction; (d) excreta and external secretions (including sweat); (e) uncannulated saliva collected either in an unstimulated fashion or stimulated by chewing gumbase or wax or by applying a dilute citric solution to the tongue; (f) placenta removed at delivery; (g) amniotic fluid obtained at the time of rupture of the membrane prior to or during labor; (h) supra- and subgingival dental plaque and calculus, provided the collection procedure is not more invasive than routine prophylactic scaling of the teeth and the process is accomplished in accordance with accepted prophylactic techniques; (i) mucosal and skin cells collected by buccal scraping or swab, skin swab, or mouth washings; (j) sputum collected after saline mist nebulization. - Collection of data through noninvasive procedures (not involving general anesthesia or sedation) routinely employed in clinical practice, excluding procedures involving x-rays or microwaves. Where medical devices are employed, they must be cleared/approved for marketing. (Studies intended to evaluate the safety and effectiveness of the medical device are not generally eligible for expedited review, including studies of cleared medical devices for new indications.)
Examples: (a) physical sensors that are applied either to the surface of the body or at a distance and do not involve input of significant amounts of energy into the subject or an invasion of the subject=s privacy; (b) weighing or testing sensory acuity; (c) magnetic resonance imaging; (d) electrocardiography, electroencephalography, thermography, detection of naturally occurring radioactivity, electroretinography, ultrasound, diagnostic infrared imaging, doppler blood flow, and echocardiography; (e) moderate exercise, muscular strength testing, body composition assessment, and flexibility testing where appropriate given the age, weight, and health of the individual. - Research involving materials (data, documents, records, or specimens) that have been collected, or will be collected solely for nonresearch purposes (such as medical treatment or diagnosis).
(NOTE: Some research in this category may be exempt from the HHS regulations for the protection of human subjects. 45 CFR 46.101 (b)-(4). This listing refers only to research that is not exempt.) - Collection of data from voice, video, digital, or image recordings made for research purposes.
- Research on individual or group characteristics or behavior (including, but not limited to, research on perception, cognition, motivation, identity, language, communication, cultural beliefs or practices, and social behavior) or research employing survey, interview, oral history, focus group, program evaluation, human factors evaluation, or quality assurance methodologies. (NOTE: Some research in this category may be exempt from the HHS regulations for the protection of human subjects. 45 CFR 46.101(b)-(2) and (b)-(3) This listing refers only to research that is not exempt.)
- Continuing review of research previously approved by the convened IRB as follows:
- where (i) the research is permanently closed to the enrollment of new subjects; (ii) all subjects have completed all research-related interventions; and (iii) the research remains active only for long-term follow-up of subjects; or
- where no subjects have been enrolled and no additional risks have been identified; or
- where the remaining research activities are limited to data analysis.
- Continuing review of research, not conducted under an investigational new drug application or investigational device exemption where categories two (2) through eight (8) do not apply but the IRB has determined and documented at a convened meeting that the research involves no greater than minimal risk and no additional risks have been identified.
In addition, expedited review is appropriate for minor changes in protocols and consent forms proposed for previously approved research during the period (one year or less) for which approval is authorized. Changes affecting risk, benefit, discomfort, or subject protections are not “minor” changes. Minor modifications include, for example, administrative changes to the protocol, changes to add follow-up calls when gathering initial data by telephone, or certain changes in the scheduling of medications.
Full IRB Review
Human subject research that does not fit any of the Expedited or Exempt review categories will require a Full IRB review at a convened meeting. The IRB meets once a month or as requested by the IRB chairman. Research applications are placed on the agenda and will be discussed at the next scheduled meeting. The IRB chairman might cancel a full IRB meeting if
- There are insufficient number of applications to be discussed,
- Inability to secure a quorum,
- University holiday; or
- Other reasons that may arise that makes a meeting unnecessary or inappropriate
The IRB uses a primary reviewer system for full IRB review. Application materials are sent to the IRB members scheduled to attend a meeting at least one week in advance of the meeting. Two members are selected by the chair, one as the primary reviewer and the other as a secondary reviewer for a research project.
The primary reviewer leads the discussion of each project at the full IRB convened meeting. The secondary reviewer adds any other relevant comments or clarifications. The members determine whether the project meets the criteria for approval or whether revisions to the study design are required. The Informed Consent Document is reviewed for accuracy, clarity, and inclusion of required and optional elements of consent. During the meeting, voting is by show of hands. By a majority of those present at the meeting, each project is either: (1) approved as submitted; (2) approved pending receipt of required minor revisions to study procedures, Informed Consent Document(s), or other written materials; (3) tabled pending review at a subsequent full board meeting after receipt of significant additional information or revisions, or (4) disapproved.
Written minutes of each full IRB meeting include: (1) attendance, (2) the number of votes to approve, table, disapprove, or abstain (without individual identification), (3) the basis for requiring changes in or disapproving the research, (4) the length of time until the next review, and (5) a summary of the discussion of controverted issues and their resolution.
Informed Consent Guidance
Informed Consent is a process in which researchers provide information to potential participants regarding the details of a research study prior to their enrollment in the study. Please refer toe the Investigator’s Manual - Policies and Procedures for a complete guidance.
Obtaining an informed consent involves:
- Providing information to the subject
- Ensuring the subject understands by answering questions the subject may have
- Obtaining voluntary agreement of the subject to participate in the study
The potential participant in a research study must participate willingly, having been adequately informed about the research. If the potential participant in a research study is part of a vulnerable population (i.e. pregnant women, cognitively impaired individuals, illiterate, children, or prisoners) special protections are required.
Special safeguards for Vulnerable Population
Special considerations must be in place to protect the rights and welfare of potential participants likely to be vulnerable to coercion or undue influence. The safeguards employed for vulnerable subjects include, among many other strategies, include the following as applicable, assessing the decision-making capacity of potential subjects, securing the involvement of a legally authorized representative, requiring parental permission from a parent/ legally authorized representative and in some studies from both parent, in addition to the child’s assent, and ensuring that incentives are not coercive.
The Informed Consent Document
The informed consent document is one aspect of the informed consent process, however it is very important. The Informed consent must
- Be written in lay terminology and at a Grade 6-8 readability level. If you are using MS Word to develop the consent document, check the readability via Flesch-Kincaid Grade Level during spell check.
- Be in a language that is understandable to the subject.
- Not contain any medical or scientific terminology. Please refer to the Glossary of Medical Lay Terminology for assistance in preparing the informed consent document.
- Not contain any abbreviations and acronyms.
- Be written in the second person: i.e.: You.
- Be free of exculpatory language.
Please refer to the checklist below as a guide while you are preparing your informed consent document. To assist the investigators in preparing the informed consent document, the ILAU RB has generated an Informed Consent Template that address the elements noted below.
Basic elements are required (Per regulations) for all informed Consent forms
- A statement that the study involves research
- An explanation of the purposes of the research
- The expected duration of the subject’s participation
- A description of the procedures to be followed
- Identification of any procedures which are experimental
- A description of any reasonably foreseeable risks or discomforts to the subject
- A description of any benefits to the subject or to others which may reasonably be expected from the research. If none, state so
- A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject
- A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained
- For research involving more than minimal risk, an explanation as to whether any compensation, and an explanation as to whether any medical treatments are available, if injury occurs and, if so, what they consist of, or where further information may be obtained
- A statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and the subject may discontinue participation at any time without penalty or loss of benefits, to which the subject is otherwise entitled
- An explanation of whom to contact for answers to pertinent questions about the research and research subjects’ rights, and whom to contact in the event of a research-related injury to the subject
- ( ) Research Qs
- ( ) Rights Qs
- ( ) Injury Qs
Additional elements as appropriate
- A statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or fetus, if the subject is or may become pregnant),
- Anticipated circumstances under which the subject’s participation may be terminated by the investigator without regard to the subject’s consent
- Any additional costs to the subject that may result from participation in the research
- The consequences of a subject’s decision to withdraw from the research and procedures for orderly termination of participation by the subject
- A statement that significant new findings developed during the course of the research, which may relate to the subject’s willingness to continue participation, will be provided to the subject
- The approximate number of subjects involved in the study
Education & Training
All investigators /student investigators and support staff involved in the research project must complete the on-line training for the Protecting of Human Research Participants.
LAU is now registered with the Collaborative Institutional Training Initiative (CITI) Program. You can access the training through the CITI Home page. Please click on “Register” and select Lebanese American University as your affiliated organization. You can refer to the following Guidance documents to complete your registration. If you are registered with CITI under a different organization, you can use the same log in information and add another affiliation under LAU.
- Humans Subject Protection training - Guidance Document related to the new training requirement and registration.
- Working with Animals training - Guidance Document related to the new training requirement and registration.
- Responsible Conduct of Research training - Guidance Document related to the new training requirement and registration. (Mandatory for Graduate students as per the Council of Deans decision (02/06/2020).
Also required for all Research Assistants and Post-Doc Research Fellows.
NOTE: All human subject training and working with animals training certification are valid for 3 years. Please make sure you re-take the training if 3 years have passed since the date on your Certificate of Completion.
Student Research and Classroom Projects
General Information
LAU students conducting research project at the Lebanese American University (LAU) or LAU Medical Center–Rizk Hospital (LAUMC–RH) must receive the necessary approval from the Institutional Review Board (IRB) at LAU as detailed below. Please read the following information carefully in order to identify which category your research falls under.
LAU students, conducting their research outside LAU or LAUMC–RH, must receive approval from the IRB at LAU as well as approval from the respective committee responsible for research involving human subjects or necessary approval where the study will be conducted.
Students are subject to the same requirements and policies set forth for the conduct of research as described below.
Definitions
Research, as defined in the Regulations - CFR Title 45, Part 46, is a systematic investigation (an organized, scientific way of collecting information, using a series of questions or observations) designed to develop or contribute to generalizable knowledge (i.e. knowledge shared by professionals in a given field which is designed to contribute to that field).
Data is collected from a “human subject”, who is a living individual about whom an investigator (a faculty member, staff or student) conducting research obtains (1) data through intervention or interaction with the individual, or (2) identifiable private information.
Above definitions are important to remember when students (undergraduate and/or graduate) are planning to conduct independent class projects, senior or honor’s theses, master’s projects, doctoral dissertations, or participate in research methodology courses. Application and submission procedures and requirements for students to follow are described in below two categories:
- Category 1 (Student Research)
- Category 2 (Classroom Projects)
Mentor / Faculty Advisor
Each student must have a mentor or a faculty advisor where the research will be taking place. The faculty advisor or mentor must be available to assist the student with developing their protocol, preparing all required documents for submission to the IRB and securing appropriate approvals prior to commencing their research.
Faculty advisors play an important role in the students’ design and development of human participant research projects. Faculty advisors are ultimately responsible for the protection of the subjects, even if the student is the primary researcher and actually directs the project, and are also responsible for research that is conducted as part of a course.
Faculty advisors and course instructors are required to review codes of ethics relevant to the discipline of study.
Categories of Research Projects by Students
Below are the two categories detailing submission requirement. Regardless which category your research project falls under, it is important to include an introductory information section prior to any data collection tool such as surveys, interview scripts, questionnaires etc. Sample introductions are available under Applications, Forms and Supporting Documents on the IRB website.
Category 1- Student Research
Student research activities include, but are not limited to, projects that result in undergraduate honors theses, masters theses, or doctoral dissertations.
Submission Requirements
Student researchers have the same submission options as any investigator. They may submit as Principal Investigator (PI) with a faculty advisor as co-signator, which may be appropriate for new projects where the student has a leading role.
Student researcher, co-investigators (if a group) and faculty advisor / classroom instructors are required to have current research ethics certification.
Common Scenarios
- RESEARCH that involves direct interaction with individuals (e.g., in person, or via mail, email, web survey, or telephone), or data from human subjects for which the researchers will have access to identifiers.
- RESEARCH-like activities using departmental subject pools (e.g., Psychology, Business, Political Science, Journalism and Mass Communication) even when the activity is conducted for educational purposes as a class requirement.
- RESEARCH that is limited to secondary analysis of data, records or specimens that are either publicly available, de-identified or otherwise impossible to be linked to personal identities
Category 2- Classroom Projects
Classroom project activities as part of Graduate and undergraduate research methodology courses: Abbreviated Review Procedure
General Guidelines
The IRB recognizes that graduate and undergraduate research methodology courses are designed to teach students research skills through a combination of readings, lectures and research activities or projects. The purpose of such research projects is for the student to apply what is being taught (i.e. use skills outside of the classroom) rather than to contribute to existing research literature in a field. Accordingly, the IRB has developed special guidelines for such class projects.
An instructor who wishes to make use of this abbreviated review procedure:
- must first diligently review each student’s proposal to determine its acceptability and then submit according to the procedures described below.
- is responsible for providing the necessary training in respecting the privacy of the individuals, the confidentiality of the data along with training in the relevant professional ethics. Such training must be listed in the course syllabus and submitted to the IRB
- Must ensure that instructor and all teaching assistants have completed the online CITI training course and have submitted their completion certificate to the IRB
Instructors are encouraged to contact the IRB for guidance about ways to handle topics such as privacy, confidentiality, informed consent, and professional ethics when class projects are part of the course syllabus. These issues may still remain even when IRB approval is not required, in which case instructors, advisors and department play an even greater role in providing the appropriate guidance and oversight.
Note: Students planning to use a class-based project as part of an undergraduate senior/honor’s thesis, master’s thesis, doctoral dissertation, independent study project, or for submitting it for off campus publication or presentation must follow the IRB review procedures before commencing the project (see Student Research Category 1 above)
Submission Requirements - Abbreviated Process
Course instructors must submit a Class Project Research Application each semester. This application will include a descriptive title of each student project, the student investigator’s name, and the type and estimated number of subjects who will be enrolled by using the Class Project Research IRB Application. The Class Project Research IRB Application form must be signed by the course instructor and submitted to the IRB Office at least one (1) week before the research is to begin. No student research project involving human subjects may begin until the instructor has submitted the application and it has been accepted by the IRB.
Common Scenarios
- CLASS PROJECTS that involve direct interaction (e.g., in person, via mail, email, web surveys, or telephone), but where the purpose is training, an educational exercise or professional development, and not research. The project is not “research” even if students ask people questions as part of learning how to conduct interviews or surveys, take histories, administer assessments, or perform “in-house” evaluations.
- Class Projects that involve direct interaction or secondary analyses of private identifiable data and are undertaken as both an educational experience and as research (e.g., results of these activities will be presented publicly or otherwise disseminated, or the data will be stored and used by the students or others as research data).
- CLASS PROJECTS involving secondary data analyses that are assigned and conducted as educational exercises, i) using data that are either publicly available data, de-identified or otherwise impossible to be linked to personal identities or ii) include private information and codes that link to identifiers, but the students do not have access to the identifiers.
Note: Projects involving but not limited to vulnerable population, collecting data sensitive in nature, collecting information in which participant might feel physically or psychologically threatened are not acceptable under this review category and a Full IRB application must be completed for review.
IRB Registration
Lebanese Ministry of Public Health
The university obtained an official authorization from the Lebanese Ministry of Public Health registering the LAU Institutional Review Board as per the Minister Decision No.141 dated 27/1/2016 under LAU IRB Authorization reference 2016/3708
To directly access information about the Laws pertaining to Clinical Trials and Authorization of IRBs, please visit the Lebanese Ministry of Public Health Website at https://www.moph.gov.lb/en/Pages/3/4760/clinical-trial-regulations
U.S Office of Human Research Protection
The university obtained a Federal Wide Assurance (FWA) as an International institution under the US Office of Human Research Protection (OHRP) which signifies that the university is in compliance with federal regulations for the protection of human subjects in research (45CFR46.103) for federally funded research. Furthermore, the university has registered its IRB, Institutional Review Board, with the US Office of Human Research Protection (OHRP).
- FWA Registration under reference FWA00014723
- IRB Registration under reference IRB00006954 Lebanese American U IRB #1
Links and References
Ethical Principles
Lebanese MOPH Regulations and CNRS Charter for Research
- Ministry of Public Health (Clinical Trials and IRB Authorization)
- Ministry of Public Health - Mental Health Program - Research in Mental Health and Psychosocial Support
- National Council for Scientific Research (CNRS) - Charter of Ethics of Scientific Research in Lebanon
Registering your Clinical Trial
European Union Clinical Trial Directive
International Conference on Harmonization
International Committee of Medical Journal Editors (ICMJE)
UNESCO
U.S Office for Human Research Protection
- Office of Human Subject Protection website
- Human Subject Protection – 45CFR46
- International Requirements for Human Subject Protection
U.S. Food and Drug Administration (FDA)
- Food and Drug Administration
- 21CFR 50 (Human Subjects)
- 21CFR 56 (IRB)
- 21 CFR 312 (IND)
- 21CFR 812 (IDE)
- Information Sheet Guidance for IRBs, Clinical Investigators and Sponsors
- A Guide to Informed Consent – Information Sheet
World Health Organization
Frequently Asked Questions
What is Research?
“Research” is defined as “a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.” The regulations further clarify that “activities which meet this definition constitute research whether or not they are conducted or supported under a program which is considered research for other purposes. For example, some demonstration and service programs may include research activities.” 45 CFR 46.102(d)
How do I know if my project involves human subjects?
“Human subjects” is defined as living individuals about whom an investigator (whether professional, faculty, staff, or student) conducting research obtains
- data through intervention or interaction with the individual, or
- identifiable private information.”
45 CFR 46.102(f)
When am I required to submit a research project involving human participants to the IRB?
All research projects that will involve human participants must be submitted for review and approval before beginning the study. This also includes proposed research involving existing data and previously collected human fluid and tissue samples, as well as any advertising or other recruitment procedures.
How long does a IRB review take?
The review varies depending on the research project. Please refer to the summary table under the Submission Requirements webpage
Who reviews my application?
The IRB reviewer depends on the type of review required.
The IRB Chair and/or an experienced member of the IRB reviews research studies that are eligible for exemption or expedited review.
For studies that require full committee review, a primary and a secondary reviewer are assigned to review the research study. A majority of the full committee (that is, a quorum) must be present at a convened meeting of the IRB in order to vote to approve or require modifications to a research study that is not exempt or eligible for expedited review.
What are the main concerns of the IRB while reviewing a research project?
The main concerns of the IRB while reviewing a research project are, but not limited to:
- Are the risks to the research subjects minimized?
- Does the study promise an acceptable risk/benefit ratio?
- Is the selection of subjects equitable? Are there appropriate justifications for the inclusion/exclusion criteria?
- How and from where will the subjects be recruited?
- Do the consent forms contain the required elements?
- Will informed consent be documented appropriately?
- Is adequate subject safety secured?
- Are there adequate provisions in place to protect subjects’ privacy and confidentiality?
- Are the routine/standard and the research procedures to be performed on subjects for the purposes of the study clearly identified?
- Are subjects informed of what is standard and what is research-related?
- Does the research plan make adequate provision for monitoring the data to ensure the safety of subjects?
- Is the purpose of the study clear and acceptable?
- Is compensation paid to subjects appropriate?
- Is there a placebo/no treatment control? If so, is the use of the placebo appropriate and does it put subjects at risk?
- Is a justification for the use of vulnerable groups provided? Does the study include additional safeguards to protect the rights and welfare of these groups?
Who is considered part of a vulnerable population?
Children, pregnant women, fetuses and neonates, the mentally disabled, individuals with diminished autonomy, prisoners, students and employees, elderly, economically disadvantaged and educationally disadvantaged.
Who needs to be listed on the protocol application?
All researchers or individuals that will have some kind of contact with the subject or with the subject’s identifiable information as follows:
- Participate in the recruitment and/or selection of participants
- Participate in the informed consent process
- Perform research related procedures
- Collect or report subject’s identifiable information (such as interviews, surveys, questionnaires, etc)
- Have access to subject’s identifiable information
What is meant by “exempt” protocol?
Under certain circumstances, human participant research activities may be granted exempt status. There are certain categories of activities that are considered research, but can be declared exempt from further review by the IRB. These categories are listed under the federal regulations, 45CFR46 101(b), and are detailed in the Protocol Exempt Application. Under exempt status, the research activity is not monitored by the IRB. Assuming the project does not change, it also is not subject to continuing IRB review and oversight. Exempt status does not however affect the ethical obligations to subjects as detailed in the Belmont Report. Thus, depending on the circumstances, investigators performing exempt studies may need to make provisions to obtain informed consent, protect confidentiality, minimize risks, and address problems or complaints.
How do I confirm that my project is exempt from IRB review?
There are certain categories of activities that are considered research, but can be declared exempt from further review by the IRB. These categories are listed under the federal regulations, 45CFR46 101(b), and are detailed in the Protocol Exempt Application. If you believe that your research project might fit one of the exempt categories, complete the Protocol Exempt Application and submit to the IRB. The IRB administrators and chairman will confirm determination and not the researcher.
I will be collaborating with another institution. Do I need to submit to the LAU IRB and the other institution?
If you are a member of the LAU faculty or staff, or an LAU student, and you are the person responsible for the conduct of the study (PI), you must get the approval from the LAU IRB to conduct your research regardless of where the research takes place. Investigators should contact the IRB office whenever collaborative research is occurring. Separate applications for each institution may be necessary.
Do I need IRB approval if my work will be conducted off campus?
All research proposals that will involve research with human subjects and will be conducted by anyone affiliated with LAU/ LAUMC–RH must be submitted to the IRB for review and approval/exemption. This requirement applies regardless of the site of the research activity.
My research involves only reviewing medical records; do I need IRB approval?
Yes, if the project meets the criteria for research with human subjects as defined above. If the review is limited to existing data, and identifiers will not be recorded, the project may qualify for exemption under category 4 (D). If prospective data collection is involved, and/or identifiers will be recorded, the protocol may be eligible for expedited review. In either case, the determination must be made by qualified IRB personnel.
My research involves administering surveys/questionnaires; do I need IRB approval? Even if I am only interviewing a few people?
Yes. Any activity conducted by an individual who is affiliated with the Lebanese American University / LAUMC–RH or conducted at LAU / LAUMC–RH that involves research on human subjects (as defined above) must be prospectively reviewed by the IRB, including those studies involving only a few subjects.
Depending upon the types of data collected, the project may be eligible for exemption or expedited review. In addition, the IRB may require additional safeguards for vulnerable populations involved in this type of research.
Do I need IRB approval to put a survey online?
Yes. Computer and internet-based research protocols must ensure voluntary participation. They address the same level of risk such as violation of privacy, legal risks, and psychosocial stress and require the same level of protection as any other types of research involving human subjects. All studies, including those using computer and internet technologies, must
- Ensure that the procedures fulfill the principles of voluntary participation and informed consent,
- Maintain the confidentiality of information obtained from or about human subjects, and
- Adequately address possible risks to subjects including psychosocial stress.
What is a continuing review?
Except for studies determined exempt from IRB oversight, all research project are required to undergo continuing review at least annually or as determined by the IRB at initial review based on the level of risk.
What do I need to do if I want to change or amend my research project?
Any changes or amendments to the protocol or any of the study documents must be submitted to the IRB for review prior to implementation.
Why is human subject training required and who must be trained?
Anyone who is listed as:
- An Investigator or “key personnel” on the IRB application
- A person obtaining informed consent,
- A contact person in the informed consent document,
- A contact person on recruitment materials for research,
- A person obtaining / reviewing individually identifiable health information
- A person conducting surveys, interviews, questionnaires, etc
Furthermore, all members of the Institutional Review Board and staff must complete the human subject training
Does the training requirement apply to students conducting interviews?
Anyone who has contact with subjects or with confidential study data must satisfy the training requirements.
Is there an expiration date on the human subject training certificate?
Yes there is an expiration date on the Certificate and it is a 3 year validity.
Contact Us
Joseph Stephan, PhD
Director of the Institutional Review Board (IRB) and Research Ethical Compliance
Email: joseph.stephan@lau.edu.lb
Ext: 2845
Gilbert and Rose-Marie Chagoury School of Medicine, Level 4
Karmen Baroudy, M.Sc.,
CIM
IRB Lead Program Manager
Email: karmen.baroudy@lau.edu.lb
Ext. 2546
IRB Office, 2nd floor Dorms A, room 704, Byblos Campus
IRB Office
Email: irb@lau.edu.lb
IRB Office, 2nd floor Dorms A, Byblos Campus